
Medizinprodukte
Medizinprodukte umfassen eine große Bandbreite von medizintechnischen Produkten und Verfahren, die Leben retten, heilen helfen und die Lebensqualität der Menschen verbessern. Nach Schätzungen des Bundesgesundheitsministeriums soll es rund 400.000 verschiedene Medizinprodukte geben. Beispiele sind Geräte für Diagnostik, Chirurgie, Intensivmedizin, Implantate, Sterilisation sowie Verbandmittel, Hilfsmittel oder OP-Material.
Definition
Als Medizinprodukt wird ein Gegenstand oder ein Stoff bezeichnet, der zu medizinisch therapeutischen oder diagnostischen Zwecken für Menschen verwendet wird, wobei die bestimmungsgemäße Hauptwirkung im Unterschied zu Arzneimitteln primär nicht pharmakologisch, metabolisch oder immunologisch, sondern physikalisch oder physikochemisch erfolgt. Die AbgrenzungExterner Link. Öffnet im neuen Fenster/Tab. der Medizinprodukte zu Arzneimitteln ist bedeutsam, da Marktzugang und Verkehrsfähigkeit unterschiedlich geregelt sind.
Die Definition ist in MDRExterner Link. Öffnet im neuen Fenster/Tab. Artikel 2 festgelegt.
Auszug aus dem Artikel 2 MDR
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands,
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper — auch aus Organ-, Blut- und Gewebespenden — stammenden Proben und dessen bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
- Produkte zur Empfängnisverhütung oder -förderung,
- Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Artikel 1 Absatz 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.
Zu beachten ist, dass auch bestimmte Produkte ohne medizinischen Verwendungszweck der MDR entsprechen müssen. Diese Produktgruppen sind in Anhang XVI MDR aufgelistet.
Klassifizierung
Die Klasse eines Medizinprodukts orientiert sich rechtlich an der "Verletzbarkeit des menschlichen Körpers" durch das jeweilige Produkt. Diese wiederum definiert sich über die Zweckbestimmung des Herstellers hinsichtlich des Anwendungsorts und der Anwendungsdauer seines Produkts. Die Medizinprodukte-Klasse bedingt den mit zunehmender Klassenhöhe ebenfalls zunehmenden Anteil an Fremdkontrolle bzw. an (externer) Zertifizierung des Konformitätsbewertungsverfahrens (Verfahren zum Nachweis der Erfüllung aller gesetzlichen Produktanforderungen) durch eine Benannte Stelle.
Kriterien für die Einteilung in 4 Klassen sind:
- Dauer der Anwendung (bis 60 Minuten, bis 30 Tage, länger als 30 Tage)
- Ort der Anwendung: Grad der Invasivität (invasiv, chirurgisch invasiv, implantierbar),
- Anwendung am zentralen Kreislaufsystem oder am zentralen Nervensystem
- Wiederverwendbares chirurgisches Instrument
- Aktives Medizinprodukt (Aktives therapeutisches Medizinprodukt / Aktives diagnostisches Medizinprodukt)
- Verwendung von biologischem Material aus Tieren oder Menschen
Die Regeln zur Klassifizierung sind detailliert im Anhang VIII der MDRExterner Link. Öffnet im neuen Fenster/Tab. festgelegt. Die Anwendung der Klassifizierungsregeln richtet sich nach der Zweckbestimmung der Produkte (und liegt daher in der Verantwortung des Herstellers).
Abgrenzung zu Arzneimitteln
Bei nichtaktiven Medizinprodukten spielt die biologische Variabilität eine geringere Rolle, da diese nicht pharmakologisch wirksam sind. Die höhere Komplexität der Wirkweise von Arzneimitteln bedeutet zudem, dass unerwünschte Arzneimittelwirkungen häufig nicht vorhersehbar sind und auch keine Aussagen über deren Eintritt, Schwere und Reversibilität gemacht werden können. Von Konstruktions- und Funktionsfehlern und einer unzureichenden Qualität abgesehen, sind unerwünschte Effekte von Medizinprodukten in stärkerem Maß vorhersehbar und aufgrund der Tatsache, dass toxische, allergene oder cancerogene Konsequenzen nicht auftreten können, in der Regel reversibel.
Weitere wesentliche Unterscheidungsmerkmale von Arzneimitteln und Medizinprodukten ergeben sich aus den Bedingungen in Bezug auf ihre Verwendung, der Anwendungsfrequenz und ihrer Funktion im Rahmen der ärztlichen Behandlung. So werden Arzneimittel vorwiegend im Rahmen der Prävention und der Therapie angewendet, während Medizinprodukte diagnostischen Zwecken dienen oder Bestandteil medizinischer Prozeduren, etwa Operationen sind. In diesem Zusammenhang stellen Arzneimittel eine eigenständige Anwendung dar, während Medizinprodukte im Rahmen von Prozessen eingesetzt werden, was dazu führt, dass deren Anteil am medizinischen Ergebnis häufig schwer zu isolieren ist. Die Effektivität von Medizinprodukten kann zudem abhängig von Faktoren sein, die nicht in einem direkten Zusammenhang mit ihrer Anwendung stehen, wozu die strukturellen und organisatorischen Rahmenbedingungen vor, während und nach der Operation zählen sind, ebenso wie die klinischen Effekte auch von den Fähigkeit und Fertigkeiten sowie der Erfahrung des Anwenders abhängig sein können.
Medizinprodukte werden von vielen kleinen und mittelständischen Unternehmen entwickelt, die nicht über ein entsprechendes Budget für eine klinische Studie verfügen. Häufig werden Medizinprodukte während der Erprobung kontinuierlich weiterentwickelt, unterliegen also kurzen iterativen Zyklen zur stetigen Qualitätsverbesserung. Anforderungen, die an Arzneimittel gestellt werden, sind deshalb nicht ohne weiteres auf Medizinprodukte übertragbar. Außerdem muss verhindert werden, dass durch zu strikte Regelungen die technische Innovation behindert wird, was letztendlich dem Patienten schaden würde.
Der Weg von der Idee zu den Patient:innen
Medizinprodukte durchlaufen umfangreiche technische Tests, bevor sie in klinischen Studien erprobt und beim Patienten angewendet werden. Neue Herzschrittmacher-Modelle werden beispielsweise über 40.000 Stunden geprüft, bis alle erforderlichen Tests durchgeführt sind. Diese Testdokumentation steht dann den Zulassungsstellen zur Verfügung. Hinzu kommt ein speziell für Medizinprodukte eingeführtes Qualitätsmanagementsystem, das Kontrollen im technischen Labor oder Chargen- und Stichprobenprüfungen umfasst, wenn die Produktion angelaufen ist.
Patientenschutz und Patientenwohl haben dabei höchste Priorität im Medizinprodukterecht. Das regulatorische System für Medizinprodukte bewährt sich seit über 15 Jahren. Medizinprodukte sind sicher, leistungsfähig und wirksam - und sie müssen dem Patienten nutzen. Hierzu gehören hohe Anforderungen:
- eine Risikoanalyse und Risikobewertung zum Nachweis der Sicherheit,
- der Nachweis der Einhaltung aller relevanten normativen und regularischen Anforderungen,
- die Durchführung einer klinischen Bewertung zum Nachweis der Leistungsfähigkeit und Wirksamkeit sowie
- ein umfassendes Qualitätsmanagementsystem.
 © BVMed
Poster: Der lange Weg eines Medizinproduktes von der Idee bis zur Anwendung an Patient:innen
Bild vergrößern
Bild herunterladen
© BVMed
Poster: Der lange Weg eines Medizinproduktes von der Idee bis zur Anwendung an Patient:innen
Bild vergrößern
Bild herunterladen
Nationales Medizinprodukterecht
Auf nationaler Ebene enthält das Medizinprodukterecht-Durchführungsgesetz (MPDG)Externer Link. Öffnet im neuen Fenster/Tab. Vorgaben zur Durchsetzung und Ergänzung der MDR. Eingeführt wurde das MPDG durch das Medizinprodukte-EU-Anpassungsgesetz (MPEUAnpG). Das MPDG gilt für Produkte im Anwendungsbereich der MDR, also für Medizinprodukte im Sinne der Verordnung. Auf In-vitro-Diagnostika ist es ab Geltungsbeginn der VO (EU) 2017/746 für In-vitro-Diagnostika (IVDR), also ab dem 26. Mai 2022 anzuwenden.
Das MPDG regelt in zehn Kapiteln und 99 Paragraphen nationale Besonderheiten. Diese betreffen beispielsweise Vorgaben für den Medizinprodukteberater, Sondervorschriften zur Durchführung klinischer Prüfungen, eine Sprachregelung für die EU-Konformitätserklärung und für Produktinformationen, Zuständigkeitsregelungen der nationalen Behörden sowie Straf- und Bußgeldvorschriften.
Ergänzt wird der Rechtsrahmen für Medizinprodukte auf nationaler Ebene daneben von folgenden Verordnungen:
- Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV)
- MPDG-Gebührenverordnung (MPDG-GebV)
- Medizinprodukte-Abgabeverordnung (MPAV)
- Medizinprodukte-Methodenbewertungsverordnung (MeMBV)
- Medizinprodukte-Betreiberverordnung (MPBetreibV)
- Apothekenbetriebsordnung (ApBetrO)
Die Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV) enthält Vorgaben zur Meldung von mutmaßlichen schwerwiegenden Vorkommnissen und regelt die Unterrichtungspflichten durch und den Informationsaustausch zwischen den Behörden. Die MPAIMV ersetzt die zuvor gültige Medizinprodukte-Sicherheitsplanverordnung (MPSV).
Die MPDG-GebV ist nur noch bis 30. September 2021 gültig. Die Gebührenerhebung im Zusammenhang mit Leistungen nach MPDG wird ab 1. Oktober 2021 in der Besonderen Gebührenverordnung des BMG gereglt sein.
Die MPAMIV und die MPDG-GebV wurden mit der Verordnung zur Anpassung des Medizinprodukterechts an die MDR und die IVDR (MPEUAnpV) eingeführt, die darüber hinaus Änderungen der MPAV, MeMBV, MPBetreibV und der ApBetrO enthielt.
Europäische Medizinprodukte-Verordnung (MDR)
Die MDRExterner Link. Öffnet im neuen Fenster/Tab. ist am 5. Mai 2017 gemeinsam mit der ebenfalls neuen Verordnung über In-vitro-Diagnostika (IVDRExterner Link. Öffnet im neuen Fenster/Tab.) im EU-Amtsblatt bekannt gemacht worden und am 25. Mai 2017 in Kraft getreten. Die MDR hatte am 26. Mai 2021 ihren Geltungsbeginn. Die bis dahin geltenden Medizinprodukterichtlinien (RL 93/42 EWG und RL 90/385/EWG) wurden mit diesem Datum aufgehoben.
Seither wurde die MDR mehrfach nachgebessert.
Mehr über die Nachbesserungen
Am 24. April 2020 wurde mit der Verordnung 2020/561Externer Link. Öffnet im neuen Fenster/Tab. der Europäischen Kommission zur Änderung der MDR unter anderem das Datum des Geltungsbeginns um ein Jahr auf den 26. Mai 2021 verschoben.
 Bild vergrößern
Bild herunterladen
Bild vergrößern
Bild herunterladen
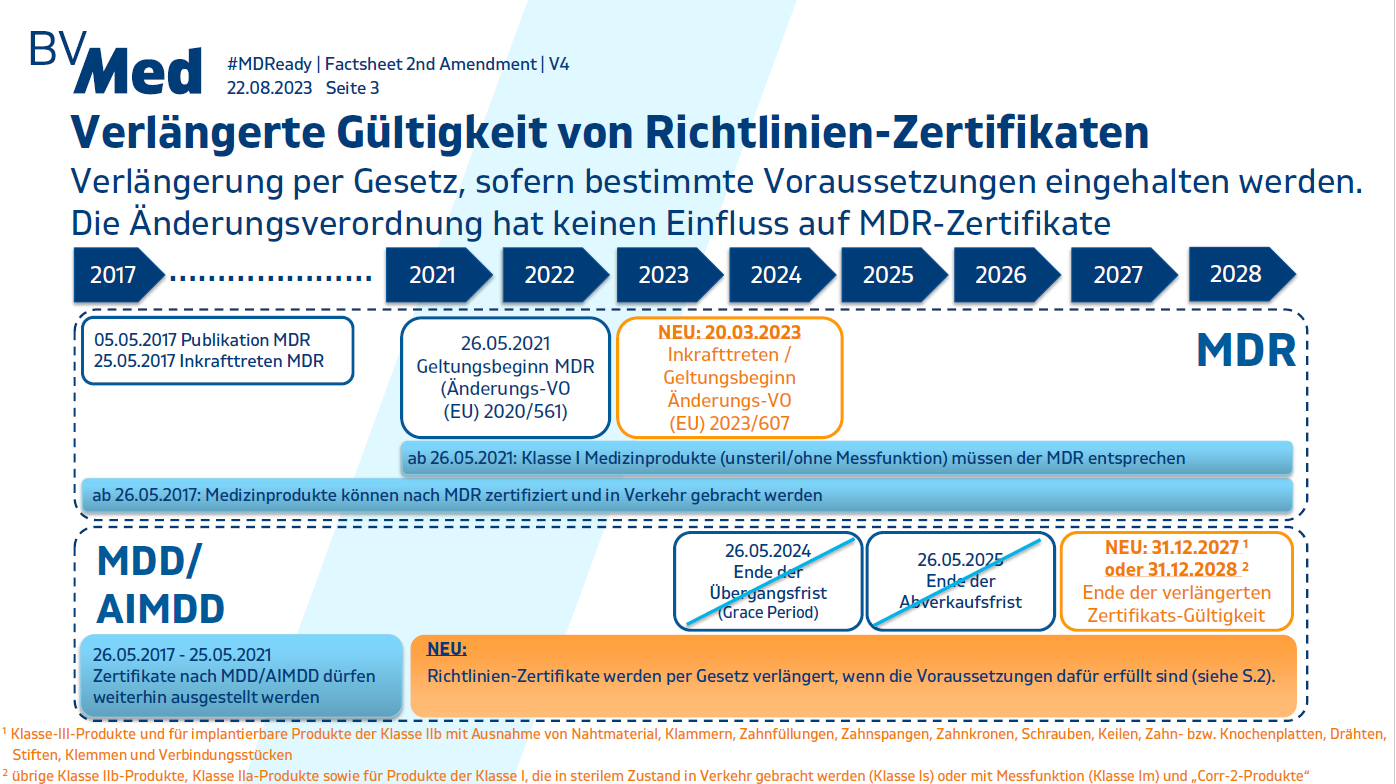
Am 20. März 2023 wurde die zweite Änderungsverordnung (VO (EU) 2023/607Externer Link. Öffnet im neuen Fenster/Tab.) publiziert, die den Kapazitätsengpass der Benannten Stellen und den Zertifikatsstau im Zuge der MDR Implementierung adressiert.
Die Änderungsverordnung (EU) 2023/607 sieht eine verlängerte Gültigkeit von Richtlinienzertifikaten, eine Verlängerung der MDR-Übergangsfristen sowie die Abschaffung der Abverkaufsfrist vor. Die Fristverlängerungen – bis 31. Dezember 2027 für Produkte der Klasse III und implantierbare Produkte der Klasse IIb sowie bis 31. Dezember 2028 für Produkte der Klassen IIa, IIb und I – sind an bestimmte Bedingungen geknüpft:
- die Produkte müssen weiterhin den MDD- bzw. AIMD-Anforderungen sowie den bisher bereits einzuhaltenden zusätzlichen MDR Anforderungen (PMS, Vigilanz, Registrierpflichten) entsprechen;
- es darf keine „wesentliche Änderung“ der Konstruktion und der Zweckbestimmung geben;
- es darf kein unannehmbares Gesundheits- oder Sicherheitsrisiko bestehen;
- der Hersteller hat spätestens am 26. Mai 2024 ein Qualitätsmanagementsystem gemäß Art. 10 Abs. 9 MDR eingerichtet;
- der Hersteller oder der/die Bevollmächtigte hat spätestens am 26. Mai 2024 bei einer Benannten Stelle einen förmlichen Antrag auf Konformitätsbewertung gestellt und die Benannte Stelle sowie der Hersteller haben bis zum 26. September 2024 eine schriftliche Vereinbarung unterzeichnet;
- das MDD-Zertifikat war am Tag des Inkrafttretens der Änderungsverordnung 2023/607 (20.03.2023) gültig. Ist dies nicht der Fall, muss der Hersteller bereits vor dem Ablauf des Zertifikats einen Vertrag mit einer Benannten Stelle über eine Konformitätsbewertung nach MDR für die Produkte abgeschlossen oder eine bestehende Ausnahme gemäß Art. 59 MDR oder Art. 97 MDR erhalten haben.
 © BVMed
#MDReady| Zeitstrahl der EU-Medizinprodukte-Verordnung MDR
Bild vergrößern
Bild herunterladen
© BVMed
#MDReady| Zeitstrahl der EU-Medizinprodukte-Verordnung MDR
Bild vergrößern
Bild herunterladen
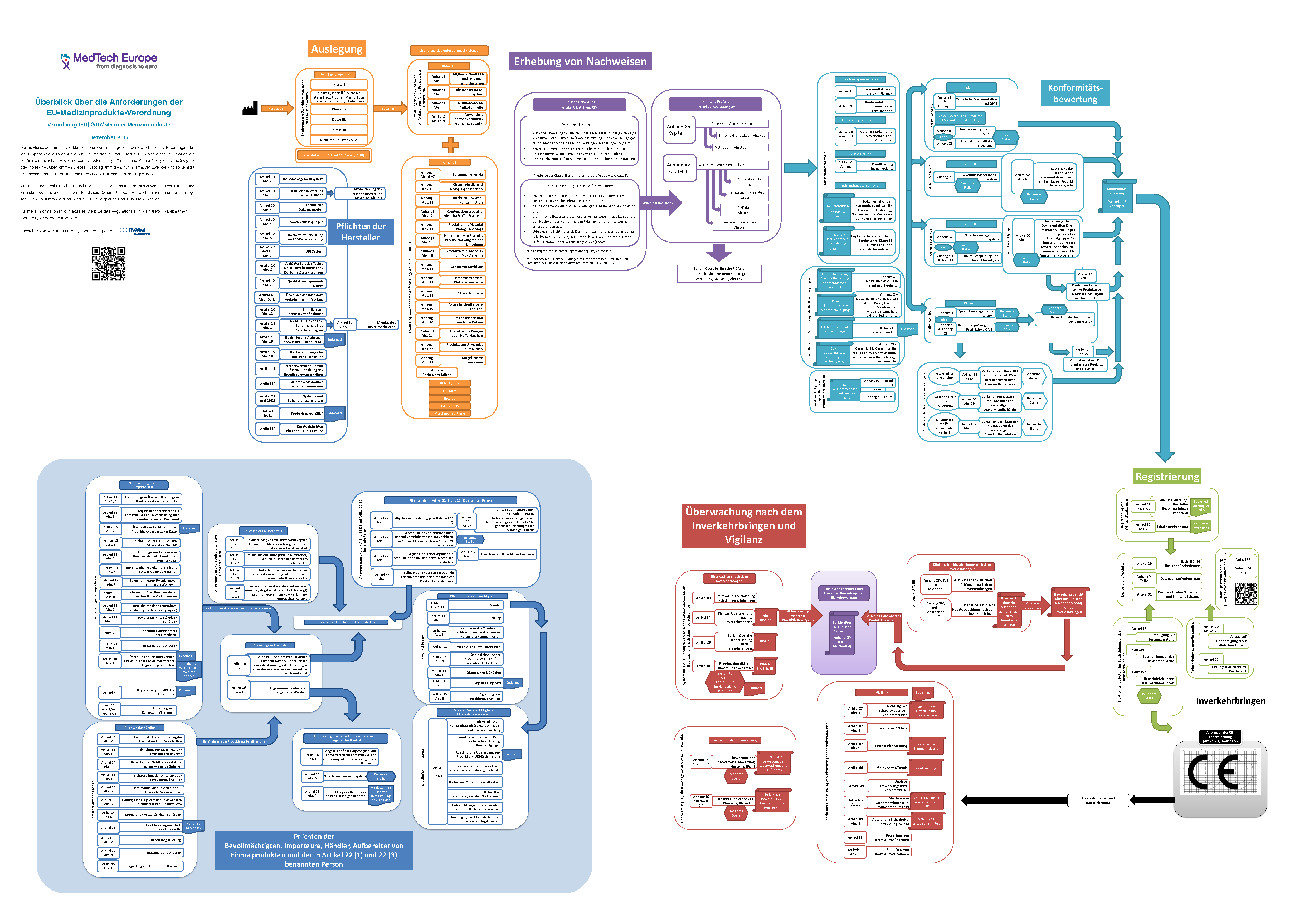
Die MDR ist ein "Basis-Rechtsakt", der über den Erlass weiterer 43 spezifischer Rechtsakte präzisiert und gesteuert wird. Darunter sind 11 delegierte und 32 durchführende Rechtsakte, von denen 8 durchführende Rechtsakte zwingend zu erlassen sind. Flowchart: Übersicht über die EU-Medizinprodukte-Verordnung (MDR)
Bild vergrößern
Bild herunterladen
Hinzu kommen europäische Leitlinien, die von der Medical Device Coordination Group als "MDCG GuidancesExterner Link. Öffnet im neuen Fenster/Tab." veröffentlicht werden.
Die MDR ist für alle Medizinprodukte seit dem 26. Mai 2021 verpflichtend anzuwenden, wobei in Art. 120 (3) MDR Übergangsfristen für Bestandsprodukte definiert sind.
In die Übergangsfristen fallen Medizinprodukte der Klasse I gemäß der Richtlinie 93/42/EWG, für die vor 26. Mai 2021 eine Konformitätserklärung erstellt wurden und für die das Konformitätsbewertungsverfahren gemäß der MDR die Mitwirkung einer Benannten Stelle erfordert sowie für Medizinprodukte höherer Klassen, für die eine Bescheinigung gemäß der Richtlinie 90/385/EWG oder der Richtlinie 93/42/EWG ausgestellt wurde, die gemäß Art. 120 Absatz 2 gültig ist.
Durch die Verordnung 2023/607 wurden die Übergangsfristen unter Bedingungen für die o.g. Produkte je nach Klasse bis Dezember 2027 bzw. Dezember 2028 verlängert. Nähere Informationen, Bedingungen und Voraussetzungen finden Sie im Merkblatt zur Inverkehrbringung von Medizinprodukten („Legacy devices“) unter der MDR nach Verordnung (EU) 2023/607 vom 20. März 2023 (2nd MDR Amendment) (Deutsch / Englisch).
Hier können Sie ein Flowchart mit einem Überblick über die MDR-Anforderungen als PDF herunterladen.







{kind=link}
{kind=link}