- Innovationen AWMF fordert ein Jahr nach Geltungsbeginn Nachbesserungen bei der Medizinprodukteverordnung MDR
ArtikelTuttlingen, 24.05.2022
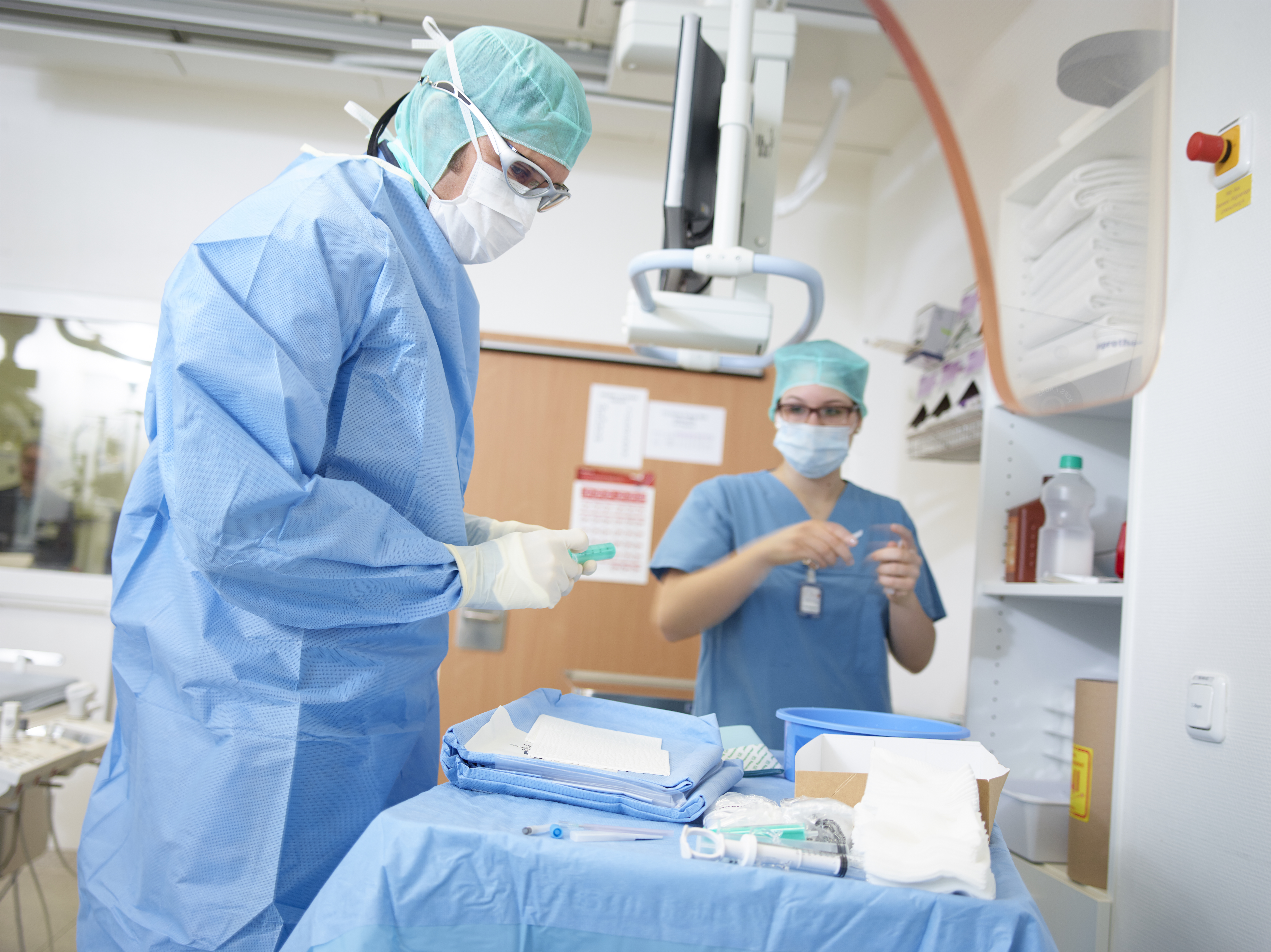
 © bvmed.de
Bild herunterladen
Die seit einem Jahr geltende europäische Verordnung für Medizinprodukte, die Medical Device Regulation (MDR), muss in der Umsetzung nachgebessert werden: Der Aufwand für Re-Zertifizierungen von bereits auf dem Markt existierenden Produkten ist zu verringern. Außerdem sollte es eine Erprobungsregelung für innovative Medizinprodukte geben, die eine schrittweise Marktzulassung ermöglicht. Das ist Thema eines Symposiums der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Fachleute aus Medizin und Technik diskutieren dort, wie die Umsetzung angelaufen ist und wie sie verbessert werden kann. Ziel der Verordnung ist es, die Sicherheit von Medizinprodukten zu erhöhen. Die regulatorischen Mehraufwände, die sich aus der Verordnung ergeben, können jedoch dazu führen, dass Hersteller von Medizinprodukten ihre Produktpallette verkleinern und dass Innovationen durch die hohen Aufwände der Zulassung gebremst werden.
© bvmed.de
Bild herunterladen
Die seit einem Jahr geltende europäische Verordnung für Medizinprodukte, die Medical Device Regulation (MDR), muss in der Umsetzung nachgebessert werden: Der Aufwand für Re-Zertifizierungen von bereits auf dem Markt existierenden Produkten ist zu verringern. Außerdem sollte es eine Erprobungsregelung für innovative Medizinprodukte geben, die eine schrittweise Marktzulassung ermöglicht. Das ist Thema eines Symposiums der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF). Fachleute aus Medizin und Technik diskutieren dort, wie die Umsetzung angelaufen ist und wie sie verbessert werden kann. Ziel der Verordnung ist es, die Sicherheit von Medizinprodukten zu erhöhen. Die regulatorischen Mehraufwände, die sich aus der Verordnung ergeben, können jedoch dazu führen, dass Hersteller von Medizinprodukten ihre Produktpallette verkleinern und dass Innovationen durch die hohen Aufwände der Zulassung gebremst werden.
Seit Mai 2021 ist die Marktzulassung für Medizinprodukte, wie beispielsweise Herzschrittmacher, Implantate, chirurgische Instrumente oder Röntgengeräte, europaweit durch die Medical Device Regulation (MDR) neu und einheitlich geregelt. Ziel war es, die Patientensicherheit weiter zu verbessern. Seitdem gehen für die Hersteller von Medizinprodukten damit beispielsweise höhere Anforderungen bei der Erstellung klinischer Daten einher. Hinzu kommen verschärfte Anforderungen bei der Zertifizierung, Re-Zertifizierung und der Nachverfolgung des Produkts über den gesamten Lebenszyklus. „Die neuen Regelungen tragen wesentlich dazu bei, die Patientensicherheit zu verbessern, was eindringlich zu begrüßen ist. Wichtig ist jedoch, dass die Umsetzung der MDR praxistauglich ist und Innovationen weiterhin möglich bleiben. In diesen beiden Bereichen zeigt sich ein Jahr nach Inkrafttreten der Neuregelung jedoch noch erheblicher Verbesserungsbedarf“, erläutert Professor Dr. med. Ernst Klar, Vorsitzender der Ad-hoc Kommission „Bewertung von Medizinprodukten“ der AWMF. „Als wissenschaftlich medizinische Fachgesellschaften wollen wir im Austausch mit allen beteiligten Einrichtungen und Verbänden dafür Sorge tragen, dass die MDR praktikabel umgesetzt wird“, ergänzt Professor Dr. Rolf-Detlef Treede, Präsident der AWMF, anlässlich des AWMF-Symposiums zur MDR im Mai 2022.
Die Medical Device Regulation sieht vor, dass auch bereits auf dem Markt verfügbare Medizinprodukte rezertifiziert werden müssen. So soll im Sinne der Patientensicherheit in Erfahrung gebracht werden, ob im Verlauf der Anwendung neue Hinweise auf Komplikationen aufgetaucht sind und der Nutzen ausreichend belegbar ist. Dies erfordert jedoch häufig klinische Daten, die bisher noch nicht erhoben wurden und auch neue klinische Studien zu den jeweiligen Produkten. „In der Praxis führt das zu erheblichen Problemen: Für die Hersteller ist der Aufwand dieser Re-Zertifizierung teilweise so hoch, dass sie das Produkt vom Markt nehmen und ihre Produktpaletten schmälern. Das wiederum kann zu Versorgungslücken führen“, betont Klar. Dies betreffe insbesondere Nischenprodukte, da sich die Aufwände durch die geringe Fertigungszahl nicht refinanzieren ließen. Für die – bisher glücklicherweise nur wenigen – Fälle, wo einzelne Medizinprodukt drohen ohne Alternative vom Markt zu verschwinden, empfiehlt die AWMF, dass das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und das Bundesministerium für Gesundheit (BMG) die Produkte begutachten und eine in der MDR vorgesehene Sonderregelung prüfen, damit die Patientenversorgung nicht gefährdet wird.
Weiteren Handlungsbedarf sieht die AWMF hinsichtlich der Benannten Stellen, welche die Produkte vor der Marktzulassung prüfen. Schätzungen zufolge müssen bis zu 55.000 Medizinprodukte rezertifiziert werden. „Die Anzahl der derzeit verfügbaren Benannten Stellen ist dafür schlicht noch nicht ausreichend“, erläutert Klar. Um eine Entlastung der Benannten Stellen zu schaffen, schlägt die AWMF eine niederschwellige Re-Zertifizierung von bereits auf dem Markt verfügbaren Medizinprodukten vor. Dafür sollten Registerdaten genutzt werden. „Anhand vorliegender Registerdaten lässt sich häufig problemlos aufzeigen, dass es in der Vergangenheit keine Sicherheitsprobleme mit dem Produkt gab und dieses eine lange Markthistorie aufweist“, betont Klar.
Das Volumen und der Aufwand der anstehenden Rezertifizierungen behindert außerdem den Marktzugang und Zertifizierung von innovativen Medizinprodukten. Die AWMF schlägt deshalb eine Erprobungsregelung vor, die helfen soll, den Marktzugang für innovative Produkte zu beschleunigen. „Die Produkte könnten so schrittweise und unter kontrollierten Bedingungen eingeführt werden. So können klinische Einrichtungen neue Medizinprodukte nicht nur in klinischen Studien testen, sondern auch mit hoher Praxisnähe in Registern verfolgen, dadurch qualifizierte Daten sammeln und die Ergebnisse später den Benannten Stellen für die weitere Zulassung zur Verfügung stellen. Die Produkte könnten anhand der in die Register gespeisten Datenpakete fortlaufend bewertet werden und ein ‚Rolling Review‘ ermöglichen. Dies bietet die Chance, dass Patientinnen und Patienten schon vor der allgemeinen Marktzulassung von innovativen Produkten profitieren könnten, selbstverständlich unter engmaschiger Kontrolle“, so Klar.
Register nehmen eine zentrale Rolle in der MDR ein. Deshalb diskutieren Expertinnen und Experten der AWMF im Rahmen ihres Symposiums auch, wie sich die Zuverlässigkeit von Registerdaten erhöhen lässt. „Hierfür ist es unabdingbar, dass es eine verpflichtende Teilnahme aller Akteure an Registern gibt. Außerdem müssen auffällige Medizinprodukte durch unabhängige Sachverständige und nicht wie bisher primär durch den Hersteller geprüft werden“, betont Professor Dr. med. Dr. med. dent. Henning Schliephake, stellvertretender Präsident der AWMF.
Die Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) e. V. bündelt die Interessen der medizinischen Wissenschaft und trägt sie verstärkt nach außen. Sie handelt dabei im Auftrag ihrer 182 medizinisch-wissenschaftlichen Fachgesellschaften. Gegründet 1962 mit dem Ziel, gemeinsame Interessen stärker gegenüber dem Staat und der ärztlichen Selbstverwaltung zu positionieren, erarbeitet die AWMF seitdem Empfehlungen und Resolutionen und vertritt diese im wissenschaftlichen und politischen Raum. Die AWMF ist Ansprechpartner für gesundheitspolitische Entscheidungsträger, wie den Gemeinsamen Bundesausschuss, und koordiniert die Entwicklung und Aktualisierung medizinisch-wissenschaftlicher Leitlinien in Deutschland. Jede gemeinnützige Fachgesellschaft in Deutschland kann Mitglied werden, sofern sie sich wissenschaftlichen Fragen der Medizin widmet. Die AWMF finanziert sich vorwiegend durch die Beiträge ihrer Mitgliedsgesellschaften und Spenden.Quelle: AWMF-Pressemeldung vom 23. Mai 2022Externer Link. Öffnet im neuen Fenster/Tab.

{kind=link}