- MDR BVMed-Konferenz zum Medizinprodukterecht: Engpässe bei den Benannten Stellen bleiben ein Problem
Die Probleme für die Hersteller von Medizinprodukten und ihre Benannten Stellen bleiben trotz der Verschiebung des Geltungsbeginns der EU-Medizinprodukte-Verordnung (MDR) auf Mai 2021 bestehen. Das verdeutlichten die Experten der BVMed-Webkonferenz „Das Medizinprodukterecht in der praktischen Umsetzung“ mit 230 Teilnehmern am 5. November 2020. Nach wie vor gibt es zu wenige Benannte Stellen: Von bislang 48 Zertifizieren sind erst 17 für die MDR notifiziert, so Martin Abel von Lohmann & Rauscher. Außerdem müssen die MDR-Audits vor Ort stattfinden, was in COVID-19-Zeiten nicht möglich ist, so Frank Matzek von Biotronik und Harald Rentschler von mdc. Dr. Nada Alkhayat von der Europäischen Kommission sieht die Verfügbarkeit der Benannten Stellen als ein „kritisches Thema“, verweist aber auf den komplexen, rund eineinhalbjährigen Benennungsprozess. Aktuell gebe es in der Kommission trotz der Engpässe keine Überlegungen, den MDR-Geltungsbeginn erneut zu verschieben, so Alkhayat. Dr. Jana Knauer vom Bundesgesundheitsministerium wies darauf hin, dass in Deutschland die Landesbehörden für Medizinprodukte zuständig bleiben, das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) „bei Gefahr im Verzug“ aber künftig selbst Maßnahmen anordnen kann.
PressemeldungBerlin, 06.11.2020, 132/20
 Dr. Martin Abel von Lohmann & Rauscher, Sprecher des BVMed-Arbeitskreises Regulatory Affairs, beleuchtete die aktuellen Herausforderungen bei der MDR-Implementierung. Zwar sei der Geltungsbeginn der MDR auf Mai 2021 verschoben worden, die Fristen für die Gültigkeit der Altzertifikate seien aber gleich geblieben. Zudem gebe es nach wie vor die Herausforderungen durch zu wenig notifizierte Benannte Stellen – aktuell sind es erst 17 von bislang 48 – sowie durch den Brexit und den „Verlust“ der größten Benannten Stelle BSI aus Großbritannien. Die MDR bringe für die Hersteller, Importeure, nationalen Vertreter oder Händler zahlreiche neue Pflichten mit sich. Das QM-System und die technische Dokumentation muss beispielsweise gemäß der MDR aufgrund neuer Hürden wesentlich aktualisiert werden. Es bedarf einer klaren „klinischen Strategie“ zum Bestehen der MDR Anforderungen und den Aufbau eines aktiven, systematischen Vigilanzsystems („Post Market Surveillance“, PMS). Durch die aktive PMS wird kontinuierlich die Anwendung, Funktionalität und Leistung des Produktes im Markt durch den Hersteller überwacht. Bei Meldungen von Vorkommnissen sind kürzere Fristen und schnellere Reaktionszeiten erforderlich. Außerdem werden die Anforderungen an die Rückverfolgbarkeit in der Vertriebskette erhöht.
Dr. Martin Abel von Lohmann & Rauscher, Sprecher des BVMed-Arbeitskreises Regulatory Affairs, beleuchtete die aktuellen Herausforderungen bei der MDR-Implementierung. Zwar sei der Geltungsbeginn der MDR auf Mai 2021 verschoben worden, die Fristen für die Gültigkeit der Altzertifikate seien aber gleich geblieben. Zudem gebe es nach wie vor die Herausforderungen durch zu wenig notifizierte Benannte Stellen – aktuell sind es erst 17 von bislang 48 – sowie durch den Brexit und den „Verlust“ der größten Benannten Stelle BSI aus Großbritannien. Die MDR bringe für die Hersteller, Importeure, nationalen Vertreter oder Händler zahlreiche neue Pflichten mit sich. Das QM-System und die technische Dokumentation muss beispielsweise gemäß der MDR aufgrund neuer Hürden wesentlich aktualisiert werden. Es bedarf einer klaren „klinischen Strategie“ zum Bestehen der MDR Anforderungen und den Aufbau eines aktiven, systematischen Vigilanzsystems („Post Market Surveillance“, PMS). Durch die aktive PMS wird kontinuierlich die Anwendung, Funktionalität und Leistung des Produktes im Markt durch den Hersteller überwacht. Bei Meldungen von Vorkommnissen sind kürzere Fristen und schnellere Reaktionszeiten erforderlich. Außerdem werden die Anforderungen an die Rückverfolgbarkeit in der Vertriebskette erhöht.
Dr. Nada Alkhayat von der Europäischen Kommission betonte, dass die COVID-19-Pandemie natürlich auch erhebliche Auswirkungen auf die Umsetzung der MDR habe. Die Kommission habe deshalb gemeinsam mit dem Parlament und dem Rat den Geltungsbeginn der MDR um ein Jahr auf Mai 2021 verschoben sowie zahlreiche „Guidances“-Dokumente zu COVDID-19 veröffentlich. Das Ende der Übergangsfrist für die Gültigkeit der Altzertifikate bleibe aber bei Mai 2024. Aktuell gebe es für den Bereich der Medizinprodukte 47 Anträge von Benannten Stellen, von denen mittlerweile 17 nach der MDR notifiziert seien. Die Kommission sei sich bewusst, dass die Verfügbarkeit der Benannten Stellen „ein kritisches Thema“ sei, ebenso wie die Verzögerungen bei Eudamed und den Expertenpanels. Hinzu kommen die fortlaufenden Einschränkungen durch COVID-19 oder die Probleme durch den Brexit sowie durch zu verhandelnde Abkommen beispielsweise mit der Schweiz.
Dr. Jana Knauer vom Referat Medizinprodukte-Sicherheit im Bundesgesundheitsministerium berichtete über die Umsetzung der MDR in nationales Recht. Die EU-Verordnung gilt unmittelbar in den Mitgliedsstaaten. Die notwendigen nationalen Regelungen wurden im Medizinprodukte-EU-Anpassungsgesetz vorgenommen. Dessen vollständiges Inkrafttreten ist aufgrund der Verschiebung der MDR ebenfalls auf den 26. Mai 2021 verschoben worden. Das nationale Medizinprodukterecht-Durchführungsgesetz (MPDG) wird damit am 26. Mai 2021 vollumfänglich in Kraft treten und das bisherige Medizinproduktegesetz (MPG) ablösen. Es wird das nationale Recht anpassen und nationale gesetzgeberische Handlungsaufträge und Möglichkeiten durch die MDR in Deutschland umsetzen. Wegen des sogenannten Wiederholungsverbots wird es allerdings kein selbst erklärendes Gesetz sein. Die Analyse des geltenden Rechts erfordert damit zukünftig die gleichzeitige Nutzung der MDR, der Leitfäden und Durchführungsakte sowie des MPDG. Im nationalen Recht bleibt es im Grundsatz bei den bisherigen Zuständigkeiten: Die Landesbehörden bleiben für Medizinprodukte zuständig. Im Bereich der Vigilanz und Marktüberwachung nimmt die Bundesoberbehörde, das BfArM, die Risikobewertung vor, die zuständige Landesbehörde trifft dann gegebenenfalls notwendige Maßnahmen. Neu ist, dass das BfArM „bei Gefahr im Verzug“ selbst Maßnahmen anordnen kann.
 Bild vergrößern
Bild herunterladen
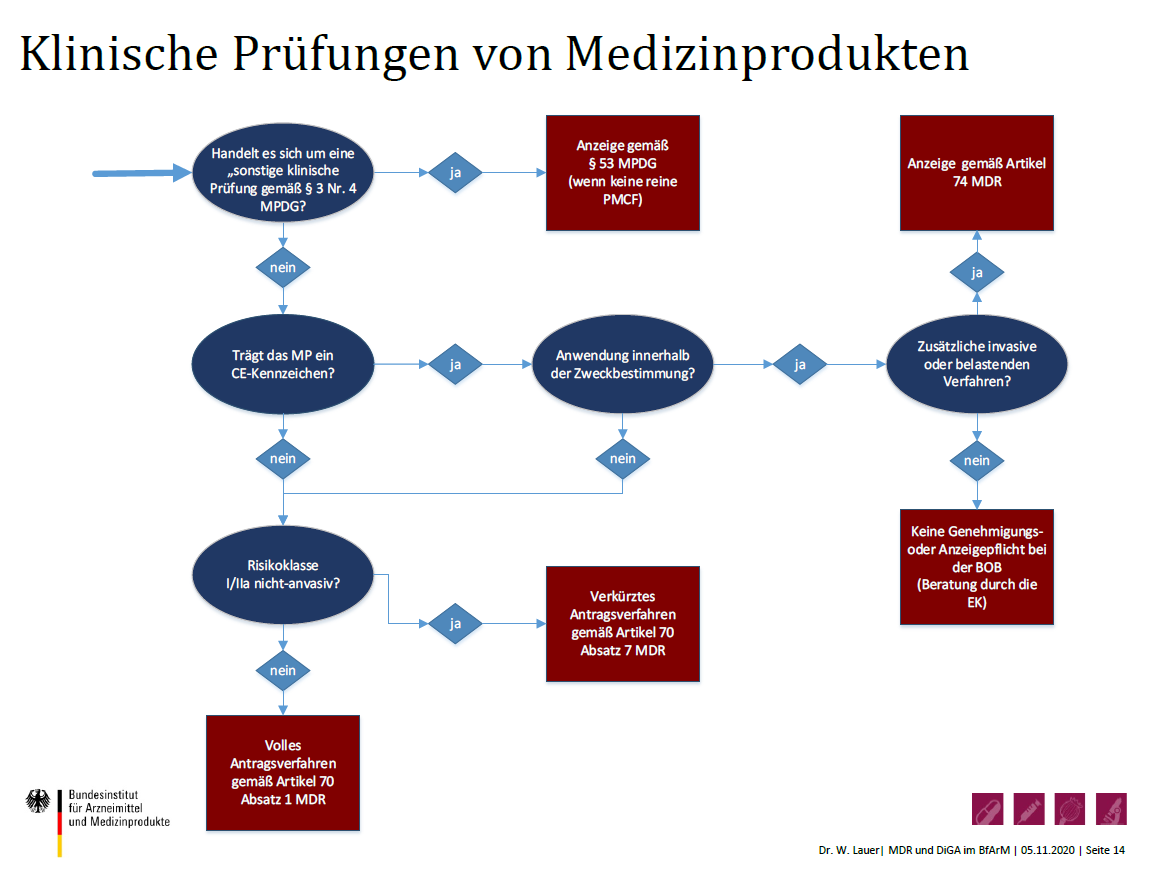
Dr. Wolfgang Lauer, Abteilungsleiter Medizinprodukte beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), ging auf die Ausnahmevorschrift der "Sonderzulassungen“ nach §7 MPDG in Verbindung mit Art. 59 MDR ein. Dies sei die einzige MDR-Vorschrift, die bereits gültig sei. Sie wurde vom BfArM in der Pandemie beispielsweise für Masken genutzt. Es handele sich aber um eine Ausnahmevorschrift „im Interesse der öffentlichen Gesundheit oder der Patientensicherheit oder -gesundheit“, nicht um eine Umgehung des regulären Verfahrens. Die Regelung diene der Sicherstellung der Verfügbarkeit von Produkten bei dringendem, alternativlosem Bedarf. Aber auch in diesen Fällen sei der Nachweis der Sicherheit und Leistungsfähigkeit erforderlich. Bei den „Klinischen Prüfungen von Medizinprodukten“ bereitet das BfArM derzeit fünf verschiedene Verfahren vor – von der Anzeige bis hin zum vollen Antragsverfahren.
Bild vergrößern
Bild herunterladen
Dr. Wolfgang Lauer, Abteilungsleiter Medizinprodukte beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), ging auf die Ausnahmevorschrift der "Sonderzulassungen“ nach §7 MPDG in Verbindung mit Art. 59 MDR ein. Dies sei die einzige MDR-Vorschrift, die bereits gültig sei. Sie wurde vom BfArM in der Pandemie beispielsweise für Masken genutzt. Es handele sich aber um eine Ausnahmevorschrift „im Interesse der öffentlichen Gesundheit oder der Patientensicherheit oder -gesundheit“, nicht um eine Umgehung des regulären Verfahrens. Die Regelung diene der Sicherstellung der Verfügbarkeit von Produkten bei dringendem, alternativlosem Bedarf. Aber auch in diesen Fällen sei der Nachweis der Sicherheit und Leistungsfähigkeit erforderlich. Bei den „Klinischen Prüfungen von Medizinprodukten“ bereitet das BfArM derzeit fünf verschiedene Verfahren vor – von der Anzeige bis hin zum vollen Antragsverfahren.
Frank Matzek vom Hersteller Biotronik kritisierte, dass die Europäische Kommission mit der Verschiebung des MDR-Geltungsbeginns die Übergangszeit für Altzertifikate bis 2024 bzw. das Ende der Abverkaufsfrist bis 2025 nicht angepasst – und damit faktisch die Übergangszeit verkürzt – hat. Die tatsächliche Übergangszeit werde zusätzlich durch die ausbleibenden MDR-Zertifizierungen während der COVID-19-Pandemie verkürzt. „Dadurch entsteht ein neuer Flaschenhals im Frühjahr 2024“, so Matzek. MDR-Erst-Zertifizierungen bzw. „MDR Scope Extensions“ sind derzeit ohne COVID-19-Bezug nicht möglich, da ein Vor-Ort-Audit erforderlich ist. Zudem verhindern fehlende Expert-Panels derzeit die Zertifizierung von einigen Hochrisikoprodukten vor dem 26. Mai 2021. Matzeks Appell: „Arbeiten sie mit ihrer Benannten Stelle an ihrem MDR-Übergangsplan und nehmen Sie Anpassungen für Ihre individuelle Situation vor. Lassen Sie sich nicht abbringen Ihre MDR-Pläne voranzutreiben.“
Harald U. Rentschler, Geschäftsführer der Benannten Stelle mdc medical device certification, berichtete, dass die erwartete Antragswelle im Bereich der Produkte der Klasse I bislang ausgeblieben sei. „Wir beobachten insgesamt eine Verzögerung der MDR-Vorbereitung bei den Herstellern. Wir haben deutlich weniger MDR-Anträge als erwartet.“ Dafür verzeichnet mdc unerwartet viele Unternehmen als Neukunden, deren bisherige Benannte Stelle weggefallen ist. Hinzu kommen sehr viele Re-Zertifizierungen nach der Medizinprodukte-Richtlinie MDD – trotz eines Zertifikatsablaufs in den Jahren 2022 oder 2023. Für die Benannten Stellen stellt der neue Benennungsprozess eine enorme Herausforderung dar. Die MDR stellt neue und detailliertere Anforderungen beispielsweise an Organisation, Ressourcen, QM-System und Zertifizierungsverfahren der Benannten Stellen. Hinzu kommen detailliertere Anforderungen an die Antragsdokumentation, ein mehrstufiges Begutachtungsverfahren einschließlich eines einwöchigen „Joint-Assessment“ durch ein internationales Team vor Ort sowie ein mehrstufiges Entscheidungsverfahren.
Der BVMed vertritt als Wirtschaftsverband über 220 Industrie- und Handelsunternehmen der Medizintechnik-Branche. Im BVMed sind u. a. die 20 weltweit größten Medizinproduktehersteller im Verbrauchsgüterbereich organisiert. Die Medizinprodukteindustrie beschäftigt in Deutschland über 215.000 Menschen und investiert rund 9 Prozent ihres Umsatzes in die Forschung und Entwicklung neuer Produkte und Verfahren. Der Gesamtumsatz der Branche liegt bei über 33 Milliarden Euro. Die Exportquote beträgt rund 65 Prozent.
{kind=link}